Research highlights
The topology of chromatin-binding domains in the NuRD deacetylase complex
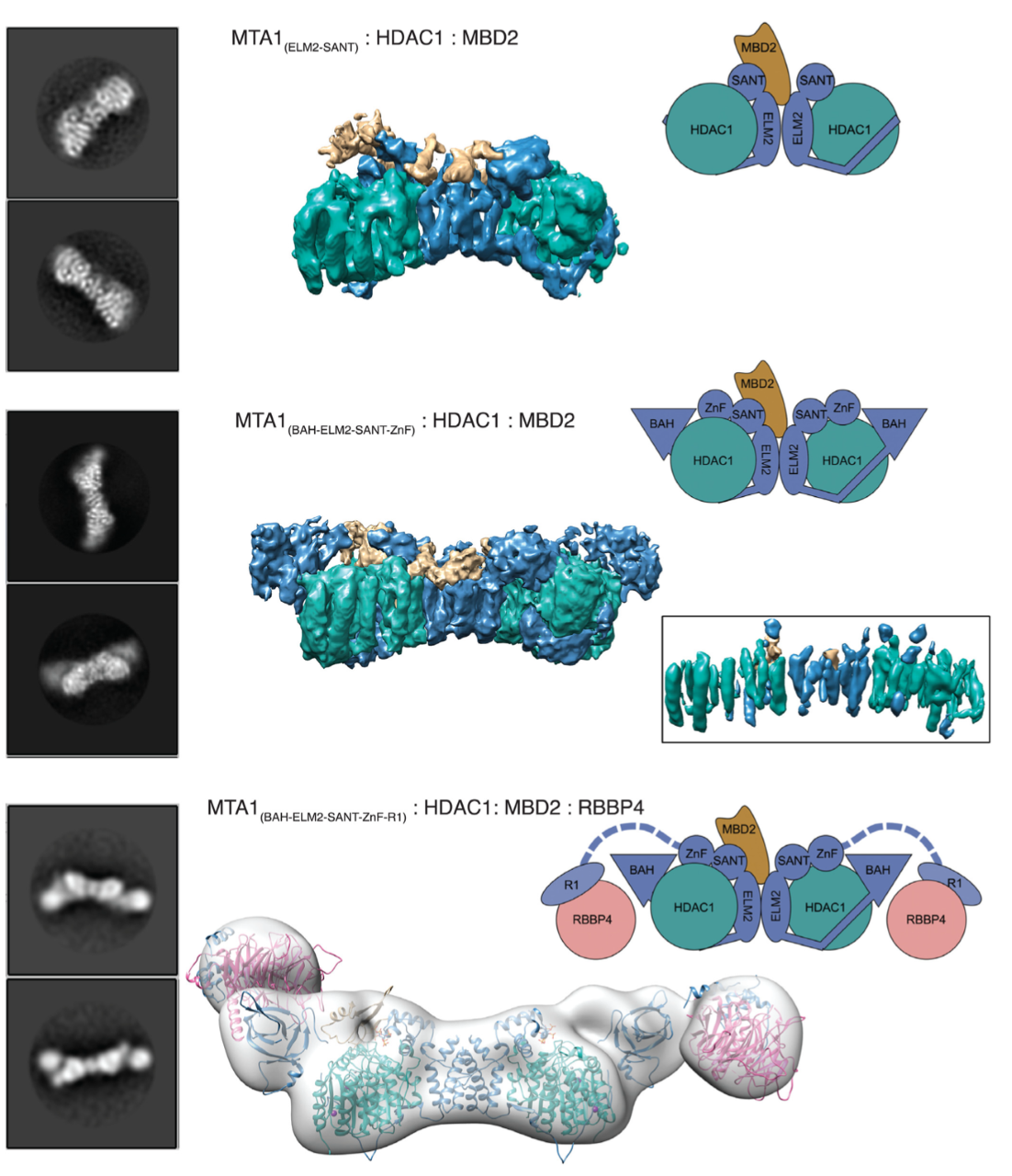
Class I histone deacetylase complexes play essential roles in many nuclear processes. Whilst they con- tain a common catalytic subunit, they have diverse modes of action determined by associated factors in the distinct complexes. The deacetylase module from the NuRD complex contains three protein do- mains that control the recruitment of chromatin to the deacetylase enzyme, HDAC1/2. Using biochem- ical approaches and cryo-electron microscopy, we have determined how three chromatin-binding do- mains (MTA1-BAH, MBD2/3 and RBBP4/7) are as- sembled in relation to the core complex so as to facil- itate interaction of the complex with the genome. We observe a striking arrangement of the BAH domains suggesting a potential mechanism for binding to di- nucleosomes. We also find that the WD40 domains from RBBP4 are linked to the core with surprising flexibility that is likely important for chromatin en- gagement. A single MBD2 protein binds asymmet- rically to the dimerisation interface of the complex. This symmetry mismatch explains the stoichiometry of the complex. Finally, our structures suggest how the holo-NuRD might assemble on a di-nucleosome substrate.
Millard, C.J., Fairall, L., Ragan, T.J., Savva, C.G. and Schwabe, J.W.R. (2020) The topology of chromatin-binding domains in the NuRD deacetylase complex. Nucleic Acids Research 48, 12972.
The MiDAC histone deacetylase complex is essential for embryonic development and has a unique multivalent structure
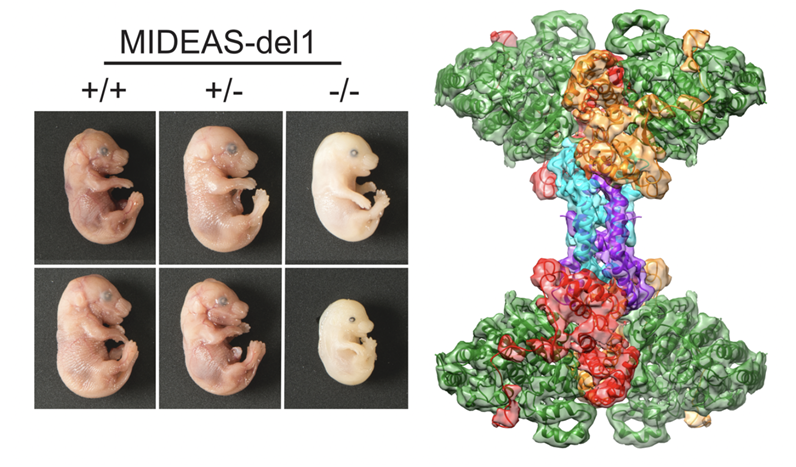
MiDAC is one of seven distinct, large multi-protein complexes that recruit class I histone deacetylases to the genome to regulate gene expression. Despite implications of involvement in cell cycle regulation and in several cancers, surprisingly little is known about the function or structure of MiDAC. Here we show that MiDAC is important for chromosome alignment during mitosis in cancer cell lines. Mice lacking the MiDAC proteins, DNTTIP1 or MIDEAS, die with identical phenotypes during late embryogenesis due to perturbations in gene expression that result in heart malformation and haematopoietic failure. This suggests that MiDAC has an essential and unique function that cannot be compensated by other HDAC complexes. Consistent with this, the cryoEM structure of MiDAC reveals a unique and distinctive mode of assembly. Four copies of HDAC1 are positioned at the periphery with outward-facing active sites suggesting that the complex may target multiple nucleosomes implying a processive deacetylase function.
- Turnbull, R., Fairall, L., Saleh, A., Kesall, E., Morris, K., Ragan, T., Savva, C., Chandru, A., Millard, C., Makarvova, O., Smith, C., Rosemann, A., Fry, A., Cowley, S., Schwabe, J.W.R. (2020) The MiDAC histone deacetylase complex is essential for embryonic development and has a unique multivalent structure. Nature Communications (2020). In Press
- Itoh, T., Fairall, L., Muskett, F. W., Milano, C. P., Watson, P. J., Arnaudo, N., Saleh, A., Millard, C.J., El-Mezgueldi, M., Martino, F., Schwabe, J.W.R. Structural and functional characterization of a cell cycle associated HDAC1/2 complex reveals the structural basis for complex assembly and nucleosome targeting. Nucleic Acids Res. (2015). doi:10.1093/nar/gkv068
Functional and structural coupling between LSD1 and HDAC1 in the CoREST complex
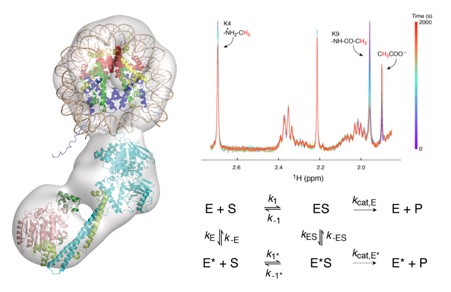 The transcriptional corepressor complex CoREST is one of seven histone deacetylase complexes that regulate the genome through controlling chromatin acetylation. The CoREST complex is unique in containing both histone demethylase and deacetylase enzymes, LSD1 and HDAC1, held together by the RCOR1 scaffold protein. To date it has been assumed that the enzymes function independently within the complex. Now, we have assembled the ternary complex. Using both structural and functional studies, we have shown that the activity of the two enzymes is closely coupled and that the complex can exist in at least two distinct states with different kinetics. Electron microscopy of the complex reveals a bi-lobed structure with LSD1 and HDAC1 enzymes at opposite ends of the complex. The structure of CoREST in complex with a nucleosome reveals a mode of chromatin engagement that contrasts with previous models.
The transcriptional corepressor complex CoREST is one of seven histone deacetylase complexes that regulate the genome through controlling chromatin acetylation. The CoREST complex is unique in containing both histone demethylase and deacetylase enzymes, LSD1 and HDAC1, held together by the RCOR1 scaffold protein. To date it has been assumed that the enzymes function independently within the complex. Now, we have assembled the ternary complex. Using both structural and functional studies, we have shown that the activity of the two enzymes is closely coupled and that the complex can exist in at least two distinct states with different kinetics. Electron microscopy of the complex reveals a bi-lobed structure with LSD1 and HDAC1 enzymes at opposite ends of the complex. The structure of CoREST in complex with a nucleosome reveals a mode of chromatin engagement that contrasts with previous models.
- Song Y., Dagil l., Fairall L., Robertson N., Wu M., Ragan T.J., Savva G.C., Morone N., Kunze M.B.A., Jamieson A.G., Cole P.A., Hansen D.F., Schwabe J.W.R. Functional and structural coupling between LSD1 and HDAC1 in the CoREST complex. Cell Reports(2020) 30, 2699-2711.
- Wu, M., Hayward, D., Kalin, J.H., Song, Y., Schwabe, J.W., Cole, P.A. Lysine-14 acetylation of histone H3 in chromatin confers resistance to the deacetylase and demethylase activities of an epigenetic silencing complex. eLife (2018) 7, e37231. doi:10.7554/eLife.3723
PROTAC-mediated degradation of class I histone deacetylase enzymes in corepressor complexes
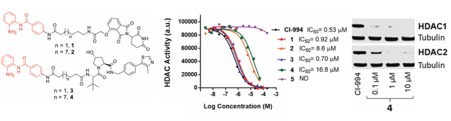 We have identified a proteolysis targeting chimera (PROTAC) of class I HDACs 1, 2 and 3. The most active degrader consists of a benzamide HDAC inhibitor, an alkyl linker, and the von Hippel-Lindau E3 ligand. Our PROTAC increased histone acetylation levels and compromised colon cancer HCT116 cell viability, establishing a degradation strategy as an alternative to class I HDAC inhibition.
We have identified a proteolysis targeting chimera (PROTAC) of class I HDACs 1, 2 and 3. The most active degrader consists of a benzamide HDAC inhibitor, an alkyl linker, and the von Hippel-Lindau E3 ligand. Our PROTAC increased histone acetylation levels and compromised colon cancer HCT116 cell viability, establishing a degradation strategy as an alternative to class I HDAC inhibition.
- Smalley, J., Adams, G., Millard, C., Song, Y., *Schwabe, J.W.R., *Cowley, S., *Hodgkinson, J. (2020) PROTAC mediated degradation of Class-I histone deacetylase enzymes in corepressor complexes. Chemical Communicationsdoi:10.1039/d0cc01485k. *Co-corresponding authors
Structure of Aster A – a novel cholesterol transport protein
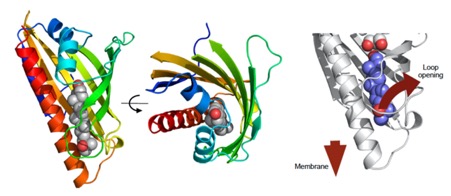
The mechanisms underlying sterol transport in mammalian cells are poorly understood. In particular, how cholesterol internalized from HDL is made available to the cell for storage or modification is unknown. Here, we describe three ER-resident proteins (Aster-A, -B, -C) that bind cholesterol and facilitate its removal from the plasma membrane. The crystal structure of the central domain of Aster-A broadly resembles the sterol-binding fold of mammalian StARD proteins, but sequence differences in the Aster pocket result in a distinct mode of ligand binding. The Aster N-terminal GRAM domain binds phosphatidylserine and mediates Aster recruitment to plasma membrane-ER contact sites in response to cholesterol accumulation in the plasma membrane. Mice lacking Aster-B are deficient in adrenal cholesterol ester storage and steroidogenesis because of an inability to transport cholesterol from SR-BI to the ER. These findings identify a nonvesicular pathway for plasma membrane to ER sterol trafficking in mammals.
- Sandhu J., Li S., Fairall L., Pfisterer S.G., Gurnett J.E., Xiao X., Weston T.A., Vashi D., Ferrari A., Orozco J.L., Hartman C.l., Strugatsky D., Lee S.D., He C., Hong C., Jiang H., Bentolila L.A., Gatta A.T., Levine T.P., Ferng A., Lee R., Ford D.A., Young S.F., *Ikonen E., *Schwabe J.W.R., *Tontonoz P. Aster proteins facilitate nonvesicular plasma membrane to ER cholesterol transport in mammalian cells. Cell (2018) 175, 1-16. *Co-senior authors
A novel dual war-head inhibitor of the CoREST complex
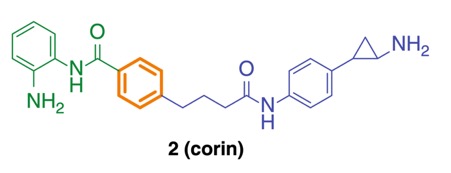
Here we report corin, a synthetic hybrid agent derived from the class I HDAC inhibitor (entinostat) and an LSD1 inhibitor (tranylcypromine analog). Enzymologic analysis reveals that corin potently targets the CoREST complex and shows more sustained inhibition of CoREST complex HDAC activity compared with entinostat. Cell-based experiments demonstrate that corin exhibits a superior anti-proliferative profile against several melanoma lines and cutaneous squamous cell carcinoma lines compared to its parent monofunctional inhibitors but is less toxic to melanocytes and keratinocytes. CoREST knockdown, gene expression, and ChIP studies suggest that corin’s favorable pharmacologic effects may rely on an intact CoREST complex. Corin was also effective in slowing tumor growth in a melanoma mouse xenograft model. These studies highlight the promise of a new class of two-pronged hybrid agents that may show preferential targeting of particular epigenetic regulatory com- plexes and offer unique therapeutic opportunities.
- Kalin, J.H., Wu, M., Gomez, A.V., Song, Y., Das, J., Hayward, D., Adejola, N., Wu, M., Panova, I., Chung, H.J., Kim, E., Roberts, H.J., Roberts, J.M., Prusevich, P., Jeliazkov, J.R., Roy Burman, S.S., Fairall, L., Milano, C., Eroglu, A., Proby, C.M., Dinkova-Kostova, A.T., Hancock, W.W., Gray, J.J., Bradner, J.E., Valente, S., Mai, A., Anders, N.M., Rudek, M.A., Hu, Y., Ryu, B., *Schwabe, J.W.R., *Mattevi, A., *Alani, R.M., *Cole, P.A. Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. Nature Communications, (2018) 9(1), 53. doi: 10.1038/s41467-017-02242-4. *Co-corresponding authors
Inositol phosphates regulate class I HDAC complexes from yeast to man
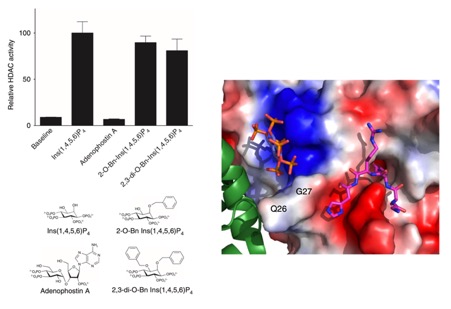
Histone deacetylases (HDACs) 1, 2 and 3 form the catalytic subunit of several large transcriptional repression complexes. Unexpectedly, the enzymatic activity of HDACs in these complexes has been shown to be regulated by inositol phosphates, which bind in a pocket sandwiched between the HDAC and co-repressor proteins. However, the actual mechanism of activation remains poorly understood. Here we have elucidated the stereochemical requirements for binding and activation by inositol phosphates, demonstrating that activation requires three adjacent phosphate groups and that other positions on the inositol ring can tolerate bulky substituents. We also demonstrate that there is allosteric communication between the inositol-binding site and the active site. The crystal structure of the HDAC1:MTA1 complex bound to a novel peptide-based inhibitor and to inositol hexaphosphate suggests a molecular basis of substrate recognition, and an entropically driven allosteric mechanism of activation.
- Watson, P.J., C.J. Millard, A.M. Riley, N.S. Robertson, L.C. Wright, H.Y. Godage, S.M. Cowley, A.G. Jamieson, B.V.L. Potter and J.W.R. Schwabe Insights into the activation mechanism of class I HDAC complexes by inositol phosphates Nature Communications(2016) 7:11262.
The architecture and function of the NuRD transcriptional repression complex
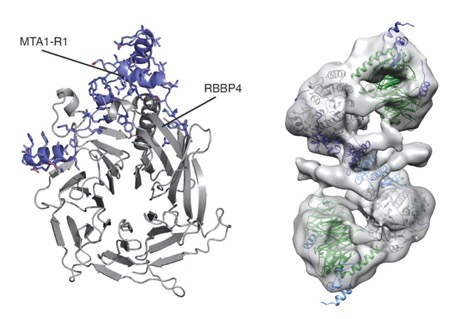
The NuRD complex is a multi-protein transcriptional corepressor that couples histone deacetylase and ATP-dependent chromatin remodelling activities. The complex regulates the higher-order structure of chromatin, and has important roles in the regulation of gene expression, DNA damage repair and cell differentiation. HDACs 1 and 2 are recruited by the MTA1 corepressor to form the catalytic core of the complex. The histone chaperone protein RBBP4, has previously been shown to bind to the carboxy-terminal tail of MTA1. We show that MTA1 recruits a second copy of RBBP4. The crystal structure reveals an extensive interface between MTA1 and RBBP4. An EM structure, supported by SAXS and crosslinking, reveals the architecture of the dimeric HDAC1: MTA1:RBBP4 assembly which forms the core of the NuRD complex. We find evidence that in this complex RBBP4 mediates interaction with histone H3 tails, but not histone H4, suggesting a mechanism for recruitment of the NuRD complex to chromatin.
- Millard, C.J., Varma, N., Saleh, A., Morris, K., Watson, P.J., Bottrill, A.R., Fairall, L., Smith, C.J. and Schwabe, J.W.R. The structure of the core NuRD complex provides insights into its interaction with chromatin eLife (2016) 5:e13941.
Human mutations in the SMRT/NCoR complex cause Pierpont syndrome and Central hypothyroidism
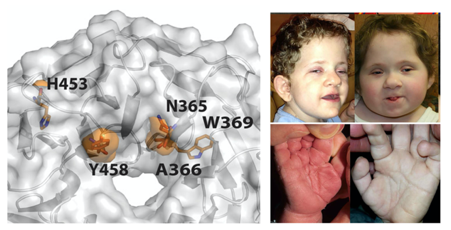
In two separate studies we have worked with clinical collaborators to understand how mutations in proteins within the SMRT/NCoR complex may result in functional defects that causing human disease. Mutations in transducin β-like 1 X-linked receptor 1 (TBL1XR1) cause Pierpont syndrome and mutations in the closely related transducin β-like protein 1, X-linked (TBL1X) cause Central hypothyroidism. Strikingly an identical mutation in both proteins (Tyrosine->Cysteine) causes both diseases. Presumably the different diseases are the consequence of perturbed function of the SMRT/NCoR complex in different tissues or at different developmental stages.
- Heinen, C.A., Jongejan, A., Watson, P.J., Redeker, B., Boelen, A., Boudzovitch-Surovtseva, O., Forzano, F., Hordijk, R., Kelley, R., Olney, A.H., Pierpont, M.E., Schaefer, B., Stewart, F., van Trotsenburg, A.S.P., Fliers, E., Schwabe, J.W.R., Hennekam, R.C. A specific mutation in TBL1XR1 causes Pierpont syndrome J Med Genet (2016) 0:1–8. doi:10.1136/jmedgenet-2015-103233.
- Heinen, C.A., Losekoot, M., Sun, Y., Watson, P.J., Fairall, L., Joustra, S.D., Zwaveling-Soonawala, N., Oostdijk, W., van den Akker, E.L.T., Alders, M., Santen, G.W.E., van Rijn, R.R., Dreschler, W.A., Surovtseva, O.V., Biermasz, N.R., Hennekam, R.C., Wit, J.M., Schwabe, J.W.R., Boelen, A., Fliers, E., Paul van Trotsenburg, A.S., (2016). Mutations in TBL1X are associated with central hypothyroidism. J Clin Endocrinol Metab 101, jc20162531–4573. doi:10.1210/jc.2016-2531
Structural and functional studies of the MiDAC complex
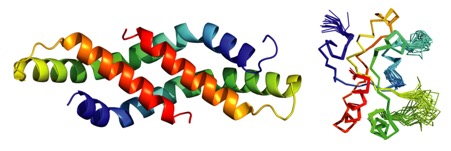 Recent proteomic studies have identified a novel histone deacetylase complex that is upregulated during mitosis and is associated with cyclin A. This complex is conserved from nematodes to man and contains histone deacetylases 1 and 2, the MIDEAS corepressor protein and a protein called DNTTIP1 whose function was hitherto poorly understood. Here we report the structures of two domains from DNTTIP1. The amino- terminal region forms a tight dimerisation domain with a novel structural fold that interacts with and mediates assembly of the HDAC1:MIDEAS complex. The carboxy- terminal domain of DNTTIP1 has a structure related to the SKI/SNO/DAC domain, despite lacking obvious sequence homology. We show that this domain in DNTTIP1 mediates interaction with both DNA and nucleosomes. Thus DNTTIP1 acts as a dimeric chromatin binding module in the HDAC1:MIDEAS corepressor complex.
Recent proteomic studies have identified a novel histone deacetylase complex that is upregulated during mitosis and is associated with cyclin A. This complex is conserved from nematodes to man and contains histone deacetylases 1 and 2, the MIDEAS corepressor protein and a protein called DNTTIP1 whose function was hitherto poorly understood. Here we report the structures of two domains from DNTTIP1. The amino- terminal region forms a tight dimerisation domain with a novel structural fold that interacts with and mediates assembly of the HDAC1:MIDEAS complex. The carboxy- terminal domain of DNTTIP1 has a structure related to the SKI/SNO/DAC domain, despite lacking obvious sequence homology. We show that this domain in DNTTIP1 mediates interaction with both DNA and nucleosomes. Thus DNTTIP1 acts as a dimeric chromatin binding module in the HDAC1:MIDEAS corepressor complex.
- Itoh, T., Fairall, L., Muskett, F. W., Milano, C. P., Watson, P. J., Arnaudo, N., Saleh, A., Millard, C.J., El-Mezgueldi, M., Martino, F., Schwabe, J.W.R. Structural and functional characterization of a cell cycle associated HDAC1/2 complex reveals the structural basis for complex assembly and nucleosome targeting. Nucleic Acids Res. (2015). doi:10.1093/nar/gkv068
The HDAC1:MTA1 dimeric complex is regulated by inositol phosphates
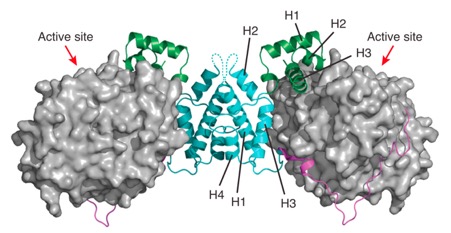
Class I histone deacetylases (HDAC1, HDAC2, and HDAC3) are recruited by cognate corepressor proteins into specific transcriptional repression complexes that target HDAC activity to chromatin resulting in chromatin condensation and transcriptional silencing. We previously reported the structure of HDAC3 in complex with the SMRT corepressor. This structure revealed the presence of inositol-tetraphosphate [Ins(1,4,5,6)P4] at the interface of the two proteins. It was previously unclear whether the role of Ins(1,4,5,6)P4 is to act as a structural cofactor or a regulator of HDAC3 activity. Here we report the structure of HDAC1 in complex with MTA1 from the NuRD complex. The ELM2-SANT domains from MTA1 wrap completely around HDAC1 occupying both sides of the active site such that the adjacent BAH domain is ideally positioned to recruit nucleosomes to the active site of the enzyme. Functional assays of both the HDAC1 and HDAC3 complexes reveal that Ins(1,4,5,6)P4 is a bona fide conserved regulator of class I HDAC complexes.
- Millard, C.J., P.J. Watson, I. Celardo, Y. Gordiyenko, S.M. Cowley, C.V. Robinson, L. Fairall, and J.W.R. Schwabe. Class I HDACs share a common mechanism of regulation by inositol phosphates. Molecular Cell (2013) 51, 57-67.
Role of inositol tetraphosphate in the HDAC3 co-repressor complex
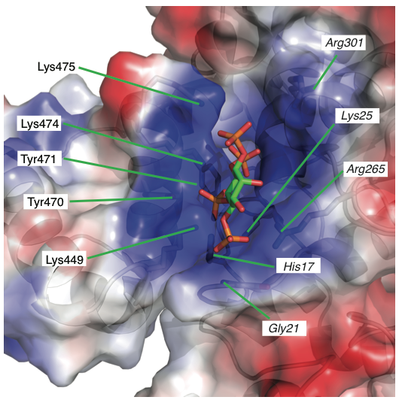
Histone deacetylase enzymes (HDACs) are emerging cancer drug targets. They regulate gene expression by removing acetyl groups from lysine residues in histone tails, resulting in chromatin condensation. The enzymatic activity of most class I HDACs requires recruitment into multi-subunit co-repressor complexes, which are in turn recruited to chromatin by repressive transcription factors. Here we report the structure of a complex between an HDAC and a co-repressor, namely, human HDAC3 with the deacetylase activation domain (DAD) from the human SMRT co-repressor (also known as NCOR2). The structure reveals two remarkable features. First, the SMRT-DAD undergoes a large structural rearrangement on forming the complex. Second, there is an essential inositol tetraphosphate molecule—D-myo-inositol-(1,4,5,6)-tetrakisphosphate (Ins(1,4,5,6)P4)—acting as an ‘intermolecular glue’ between the two proteins. Assembly of the complex is clearly dependent on the Ins(1,4,5,6)P4, which may act as a regulator—potentially explaining why inositol phosphates and their kinases have been found to act as transcriptional regulators. This mechanism for the activation of HDAC3 appears to be conserved in class I HDACs from yeast to humans, and opens the way to novel therapeutic opportunities.
- Watson, P.J., L. Fairall, G.M. Santos and J.W.R. Schwabe. Structure of HDAC3 bound to corepressor and inositol tetraphosphate. Nature 2012, 481 p: 335-340.
Understanding how the E3 ligase IDOL targets the LDL receptor for degradation
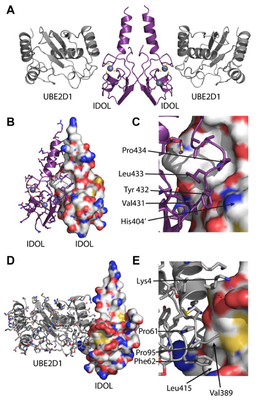
We previously identified the E3 ubiquitin ligase IDOL as a sterol-dependent regulator of the LDL receptor (LDLR). The molecular pathway underlying IDOL action, however, remains to be determined. Here we report the identification and biochemical and structural characterization of an E2–E3 ubiquitin ligase complex for LDLR degradation. We identified the UBE2D family (UBE2D1–4) as E2 partners for IDOL that support both autoubiquitination and IDOL-dependent ubiquitination of the LDLR in a cell-free system. NMR chemical shift mapping and a 2.1 A ̊ crystal structure of the IDOL RING domain–UBE2D1 complex revealed key interactions between the dimeric IDOL protein and the E2 enzyme. Analysis of the IDOL–UBE2D1 interface also defined the stereochemical basis for the selectivity of IDOL for UBE2Ds over other E2 ligases. Structure-based mutations that inhibit IDOL dimerization or IDOL–UBE2D interaction block IDOL-dependent LDLR ubiquitination and degradation. Furthermore, expression of a dominant-negative UBE2D enzyme inhibits the ability of IDOL to degrade the LDLR in cells. These results identify the IDOL–UBE2D complex as an important determinant of LDLR activity, and provide insight into molecular mechanisms underlying the regulation of cholesterol uptake.
- Zhang, L., L. Fairall, B.T. Goult, A.C. Calkin, C. Hong, C.J. Millard, P. Tontonoz, and J.W.R. Schwabe, The IDOL-UBE2D complex mediates sterol-dependent degradation of the LDL receptor. Genes Dev 2011, 25 p: 1262-1274.
The E3 ubiquitin ligase IDOL (inducible degrader of the LDL receptor) regulates LDL receptor (LDLR)-dependent cholesterol uptake, but its mechanism of action, including the molecular basis for its stringent specificity, is poorly understood. Here we show that IDOL uses a singular strategy among E3 ligases for target recognition. The IDOL FERM domain binds directly to a recognition sequence in the cy- toplasmic tails of lipoprotein receptors. This physical interaction is independent of IDOL’s really interesting new gene (RING) domain E3 ligase activity and its capacity for autoubiquitination. Furthermore, IDOL controls its own stability through autoubiquitination of a unique FERM subdomain fold not present in other FERM proteins. Key resi- dues defining the IDOL–LDLR interaction and IDOL autoubiquitina- tion are functionally conserved in their insect homologs. Finally, we demonstrate that target recognition by IDOL involves a tripartite in- teraction between the FERM domain, membrane phospholipids, and the lipoprotein receptor tail. Our data identify the IDOL–LDLR inter- action as an evolutionarily conserved mechanism for the regulation of lipid uptake and suggest that this interaction could potentially be exploited for the pharmacologic modulation of lipid metabolism.
- Calkin, A, B.T. Goult, L. Zhang, L. Fairall, C. Hong, J.W.R. Schwabe* and P. Tontonoz*. FERM- dependent E3 ligase recognition: an evolutionarily-conserved mechanism for targeted degradation of lipoprotein receptors. Proc Natl Acad Sci U S A 2011, 108 p: 20107-20112. *Co-corresponding authors
Assembly of the SMRT/NCoR transcriptional repression complex
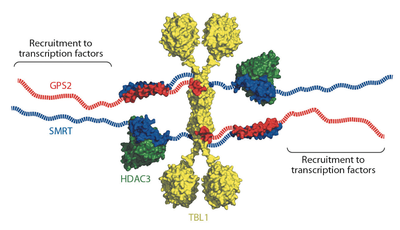
Eukaryotic transcriptional repressors function by recruiting large coregulatory complexes that target histone deacetylase enzymes to gene promoters and enhancers. Transcriptional repression complexes, assembled by the corepressor NCoR and its homolog SMRT, are crucial in many processes, including development and metabolic physiology. The core repression complex involves the recruitment of three proteins, HDAC3, GPS2 and TBL1, to a highly conserved repression domain within SMRT and NCoR. We have used structural and functional approaches to gain insight into the architecture and biological role of this complex. We report the crystal structure of the tetrameric oligomerization domain of TBL1, which interacts with both SMRT and GPS2, and the NMR structure of the interface complex between GPS2 and SMRT. These structures, together with computational docking, mutagenesis and functional assays, reveal the assembly mechanism and stoichiometry of the corepressor complex.
- Oberoi, J., L. Fairall, P. Watson, J.-C. Yang, Z. Czimmerer, T. Kampmann, B. Goult, J. Greenwood, J. Gooch, B.C. Kallenberger, L. Nagy, D. Neuhaus, and J.W.R. Schwabe, Structural basis for the assembly of the SMRT/NCoR core transcriptional repression machinery. Nat Struct Mol Biol 2011, 18 p: 177-84.
The activation of PPARgamma by oxidised fatty acids
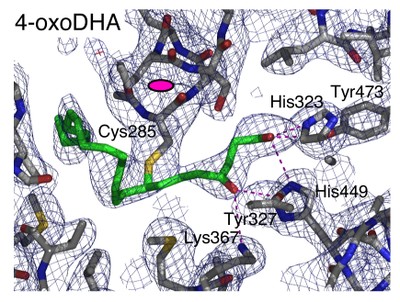
The nuclear receptor PPARgamma has important roles in adipogenesis and immune response as well as roles in both lipid and carbohydrate metabolism. Although synthetic agonists for PPARgamma are widely used as insulin sensitisers, the identity of the natural ligand(s) for PPARgamma is still not clear. Suggested natural ligands include 15-deoxy-D12,14-prostaglandin J2 and oxidised fatty acids such as 9-HODE and 13-HODE. Crystal structures of PPARgamma have revealed the mode of recognition for synthetic compounds.
Here we report structures of PPARgamma bound to oxidised fatty acids that are likely to be natural ligands for this receptor. These structures reveal that the receptor can (i) simultaneously bind two fatty acids and (ii) couple covalently with conjugated oxo fatty acids. Thermal stability and gene expression analyses suggest that such covalent ligands are particularly effective activators of PPARgamma and thus may serve as potent and biologically relevant ligands.
- Itoh T, Fairall L, Amin K, Inaba Y, Szanto A, Balint BL, Nagy L, Yamamoto K, Schwabe JWR. Structural basis for the activation of PPARgamma by oxidised fatty acids. Nat Struct Mol Biol 2008, 15: p. 924-931.
The histone deacetylase activation domain from SMRT
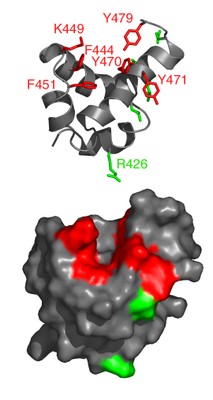
SMRT (silencing mediator of retinoid acid and thyroid hormone receptor) and NCoR (nuclear receptor corepressor) are transcriptional corepressors that play an essential role in the regulation of development and metabolism.
This role is achieved, in part, through the recruitment of a key histone deacetylase (HDAC3), which is itself indispensable for cell viability.
The assembly of HDAC3 with the deacetylase activation domain (DAD) of SMRT and NCoR is required for activation of the otherwise inert deacetylase. The DAD comprises an N-terminal DAD-specific motif and a C-terminal SANT (SWI3_ADA2_NCoR_TFIIIB)-like domain. We report here the solution structure of the DAD from SMRT, which reveals a four-helical structure. The DAD differs from the SANT (and MYB) domains in that (i) it has an additional N-terminal helix and (ii) there is a notable hydrophobic groove on the surface of the domain.
Structure-guided mutagenesis, combined with interaction assays, showed that residues in the vicinity of the hydrophobic groove are required for interaction with (and hence activation of) HDAC3. Importantly, one surface-exposed lysine is required for activation of HDAC3, but not for interaction. This lysine may play a uniquely important role in the mechanism of activating HDAC3.
- Codina A, Love JD, Li Y, Lazar MA, Neuhaus D, Schwabe JWR. Structural insights into the interaction and activation of HDAC3 by nuclear receptor co-repressors. Proc Natl Acad Sci USA 2005, 102: p. 6009-6014.
Novel co-regulator binding surface on Nurr1
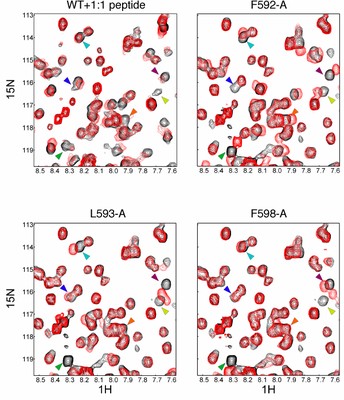
The nuclear receptor Nurr1 is a transcription factor essential for the development of midbrain dopaminergic neurons in vertebrates. Recent crystal structures of the Nurr1 ligand binding domain (LBD) and the Drosophila orthologue dHR38 revealed that, although these receptors share the classical LBD architecture, they lack a ligand binding cavity.
This volume is instead filled with bulky hydrophobic side chains. Furthermore the 'canonical' non-polar co-regulator binding groove is filled with polar side chains; thus, the regulation of transcription by this sub-family of nuclear receptor LBDs may be mediated by some other interaction surface on the LBD.
We report here the identification of a novel co-regulator interface on the LBD of Nurr1. We used an NMR footprinting strategy that facilitates the identification of an interaction surface without the need of a full assignment. We found that non-polar peptides derived from the co-repressors SMRT and NCoR bind to a hydrophobic patch on the LBD of Nurr1. This binding surface involves a groove between helices 11 and 12. Mutations in this site abolish activation by the Nurr1 LBD. These findings give insight into the unique mechanism of action of this class of nuclear receptors.
- Codina A, Benoit G, Gooch JT, Neuhaus D, Perlmann T, Schwabe JW. Identification of a novel co-regulator interaction surface on the ligand binding domain of Nurr1 using NMR footprinting. J Biol Chem 2004, 279: p. 53338-45.
The SPOC Domain from SHARP
Spen proteins regulate the expression of key transcriptional effectors in diverse signaling pathways. They are large proteins characterised by N-terminal RNA-binding motifs and a highly conserved C-terminal SPOC domain.
The specific biological role of the SPOC domain (Spen paralog and ortholog C-terminal domain), and hence the common function of Spen proteins, has been unclear to date. The Spen protein SHARP (SMRT/HDAC1-associated repressor protein) was identified as a component of transcriptional repression complexes in both nuclear receptor and Notch/RBP-Jkappa signaling pathways. We have determined the 1.8 A crystal structure of the SPOC domain from SHARP. This structure shows that essentially all of the conserved surface residues map to a positively charged patch.
Structure-based mutational analysis indicates that this conserved region is responsible for the interaction between SHARP and the universal transcriptional corepressor SMRT/NCoR (silencing mediator for retinoid and thyroid receptors/nuclear receptor corepressor). We demonstrate that this interaction involves a highly conserved acidic motif at the C terminus of SMRT/NCoR. These findings suggest that the conserved function of the SPOC domain is to mediate interaction with SMRT/NCoR corepressors and that Spen proteins play an essential role in the repression complex.
- Ariyoshi M, Schwabe JW. A conserved structural motif reveals the essential transcriptional repression function of Spen proteins and their role in developmental signaling. Genes Dev, 2003. 17: p. 1909-20.
Nuclear receptor dynamics
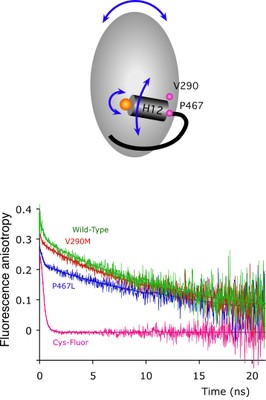
Nuclear receptors are transcription factors that activate gene expression in response to ligands. The C-terminal helix (helix 12) of the ligand-binding domain plays a critical role in the activation mechanism. When bound to activating ligands, helix 12 adopts a conformation that promotes the binding of co-activator proteins. Helix 12 also adopts this 'active' position in several ligand-free structures, raising questions as to the exact role of helix 12.
We proposed that the dynamic properties of helix 12 may be critical for the activation mechanism and, to test this, have used fluorescence anisotropy techniques to directly monitor the mobility of helix 12 in PPARgamma. Our results suggest that helix 12 is significantly more mobile than the main body of the protein. Upon ligand binding, helix 12 shows reduced mobility, accounting for its role as a molecular switch.
We also show that natural mutations in human PPARgamma, associated with severe insulin resistance and diabetes mellitus, exhibit perturbations in the dynamic behavior of helix 12. Our findings provide the first direct observations of the mobility of helix 12 and suggest that the dynamic properties of this helix are key to the regulation of transcriptional activity.
- Kallenberger BC, Love JD, Chatterjee VK, Schwabe JW. A dynamic mechanism of nuclear receptor activation and its perturbation in a human disease. Nat Struct Biol, 2003. 10: p. 136-40.
Specific ligands for RXR
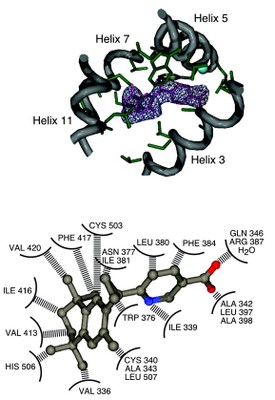
Ligands that specifically target retinoid-X receptors (RXRs) are emerging as potentially powerful therapies for cancer, diabetes,and the lowering of circulatory cholesterol. To date, RXR has only been crystallised in the absence of ligand or with the promiscuous ligand 9-cis retinoic acid, which also activates retinoic acid receptors.
Here we present the structure of hRXRbeta in complex with the RXR-specific agonist LG100268 (LG268). The structure clearly reveals why LG268 is specific for the RXR ligand binding pocket and will not activate retinoic acid receptors.
Intriguingly, in the crystals, the C-terminal 'activation' helix (AF-2/helix H12) is trapped in a novel position not seen in other nuclear receptor structures such that it does not cap the ligand binding cavity. Mammalian two-hybrid assays indicate that LG268 is unable to release co-repressors from RXR unless co-activators are also present. Together these findings suggest that RXR ligands may be inefficient at repositioning helix H12.
- Love JD, Gooch JT, Benko S, Li C, Nagy L, Chatterjee VK, Evans RM, Schwabe JW. The structural basis for the specificity of retinoid-X receptor-selective agonists: new insights into the role of helix H12. J Biol Chem, 2002. 277: p. 11385-91.
An unexpected ligand for Ultraspiracle
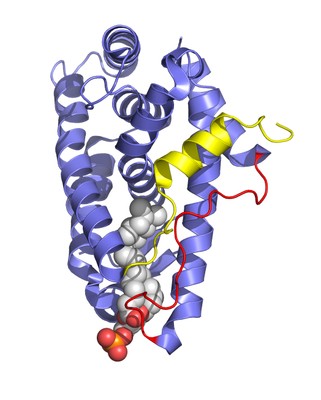
Ultraspiracle (USP) is the invertebrate homologue of the mammalian retinoid X receptor (RXR). RXR plays a uniquely important role in differentiation, development, and homeostasis through its ability to serve as a heterodimeric partner to many other nuclear receptors.
RXR is able to influence the activity of its partner receptors through the action of the ligand 9-cis retinoic acid. In contrast to RXR, USP has no known high-affinity ligand and is thought to be a silent component in the heterodimeric complex with partner receptors such as the ecdysone receptor.
Here we report the 2.4-Å crystal structure of the USP ligand-binding domain. The structure shows that a conserved sequence motif found in dipteran and lepidopteran USPs, but not in mammalian RXRs, serves to lock USP in an inactive conformation. It also shows that USP has a large hydrophobic cavity, implying that there is almost certainly a natural ligand for USP. This cavity is larger than that seen previously for most other nuclear receptors. Intriguingly, this cavity has partial occupancy by a bound lipid, which is likely to resemble the natural ligand for USP.
- Clayton GM, Peak-Chew SY, Evans RM, Schwabe JW. The structure of the ultraspiracle ligand-binding domain reveals a nuclear receptor locked in an inactive conformation. Proc Natl Acad Sci USA, 2001. 98: p. 1549-54.
How co-repressors bind nuclear receptors
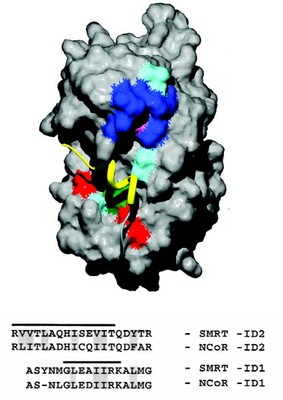
The association of transcription corepressors SMRT and N-CoR with retinoid and thyroid receptors results in suppression of basal transcriptional activity. A key event in nuclear receptor signaling is the hormone-dependent release of corepressor and the recruitment of coactivator. Biochemical and structural studies have identified a universal motif in coactivator proteins that mediates association with receptor LBDs.
We report here the identity of complementary acting signature motifs in SMRT and N-CoR that are sufficient for receptor binding and ligand-induced release. Interestingly, the motif contains a hydrophobic core (PhixxPhiPhi) similar to that found in NR coactivators. Surprisingly, mutations in the amino acids that directly participate in coactivator binding disrupt the corepressor association. These results indicate a direct mechanistic link between activation and repression via competition for a common or at least partially overlapping binding site.
- Nagy L, Kao HY, Love JD, Li C, Banayo E, Gooch JT, Chatterjee VKK, Evans RM, Schwabe JW. Mechanism of corepressor binding and release from nuclear hormone receptors. Genes Dev, 1999. 13: p. 3209-16.
- Benko S, Love JD, Beládi M, Schwabe, JWR, Nagy L. Molecular determinants of the balance between co-repressor and co-activator recruitment to the retinoic acid receptor: identification of residues that bias helix 12 positioning. J. Biol. Chem., 2003. 278: p. 43797-806.
DNA-binding by the oestrogen receptor
The nuclear hormone receptors are a superfamily of ligand-activated DNA-binding transcription factors. We have determined the crystal structure (at 2.4Å) of the fully specific complex between the DNA-binding domain from the estrogen receptor and DNA.
The protein binds as a symmetrical dimer to its palindromic binding site consisting of two 6 bp consensus half sites with three intervening base pairs. This structure reveals how the protein recognises its own half site sequence rather than that of the related glucocorticoid receptor, which differs by only two base pairs. Since all nuclear hormone receptors recognise one or the other of these two consensus half site sequences, this recognition mechanism applies generally to the whole receptor family.
- Schwabe JW, Chapman L, Finch JT, Rhodes D. The crystal structure of the estrogen receptor DNA-binding domain bound to DNA: how receptors discriminate between their response elements. Cell, 1993. 75: p. 567-78.